
PathIntegrate: Multivariate Modelling Approaches for Pathway-Based Multi-Omics Data Integration
Published in PLOS Computational Biology, 2024
Unsupervised Extension to the PathIntegrate Machine Learning Class:
Incorporated two new functions into the package:
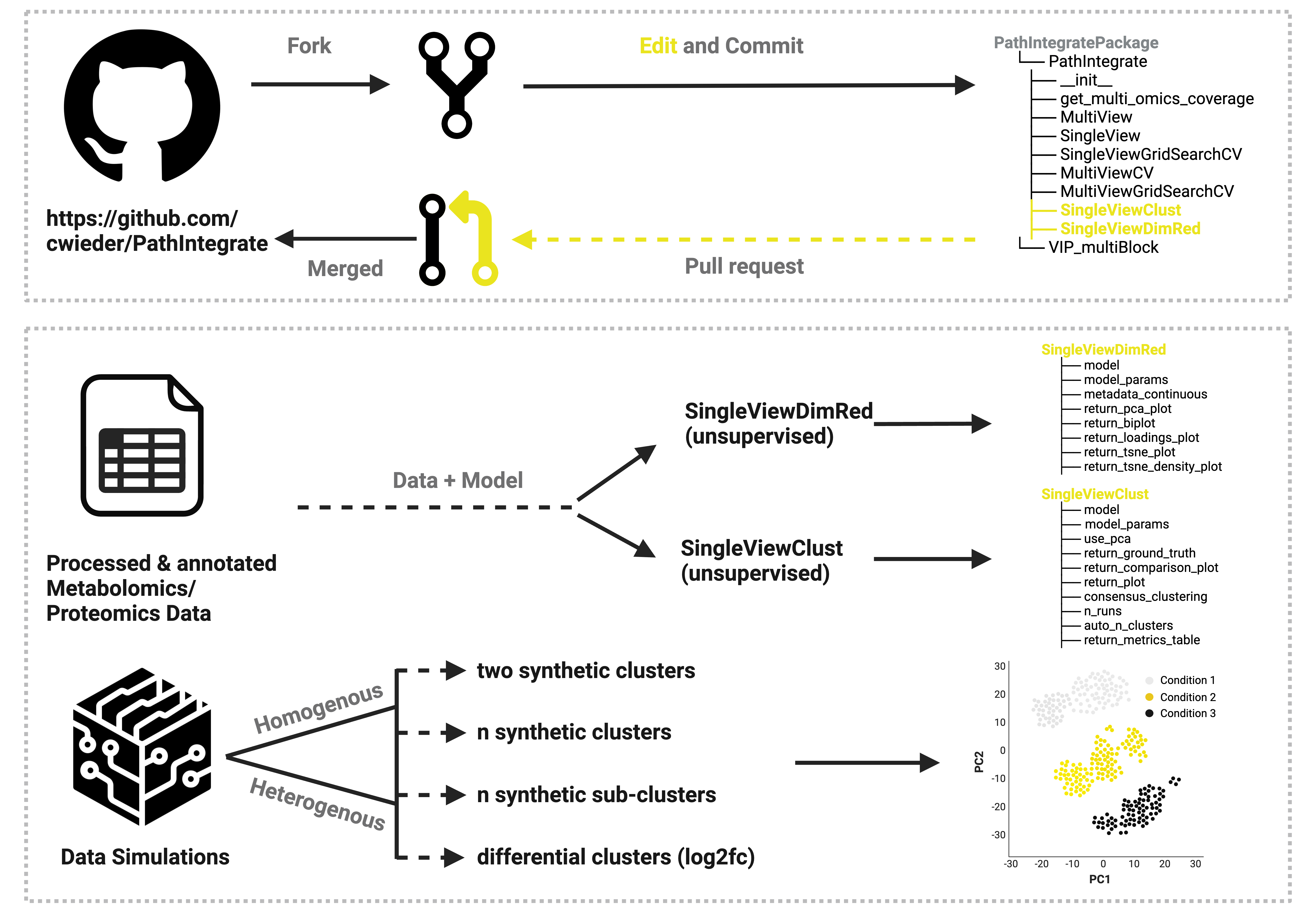
Tutorial:
Please visit: https://github.com/judepops/MultiomicsML
The tutorial is detailed in the ReadMe
Code:
import pandas as pd import numpy as np import sklearn.decomposition import sspa import sklearn from mbpls.mbpls import MBPLS from pathintegrate.app import launch_network_app from sklearn.preprocessing import StandardScaler from sklearn.pipeline import Pipeline from sklearn.model_selection import cross_val_score, GridSearchCV import scipy import seaborn as sns import matplotlib.pyplot as plt import requests import json from sklearn.metrics import confusion_matrix import plotly.graph_objects as go from io import BytesIO import base64
class PathIntegrate: ‘'’PathIntegrate class for multi-omics pathway integration.
Args:
omics_data (dict): Dictionary of omics data. Keys are omics names, values are pandas DataFrames containing omics data where rows contain samples and columns reprsent features.
metadata (pandas.Series): Metadata for samples. Index is sample names, values are class labels.
pathway_source (pandas.DataFrame): GMT style pathway source data. Must contain column 'Pathway_name'.
sspa_scoring (object, optional): Scoring method for ssPA. Defaults to sspa.sspa_SVD. Options are sspa.sspa_SVD, sspa.sspa_ssGSEA, sspa.sspa_KPCA, sspa.sspa_ssClustPA, sspa.sspa_zscore.
min_coverage (int, optional): Minimum number of molecules required in a pathway. Defaults to 3.
Attributes:
omics_data (dict): Dictionary of omics data. Keys are omics names, values are pandas DataFrames.
omics_data_scaled (dict): Dictionary of omics data scaled to mean 0 and unit variance. Keys are omics names, values are pandas DataFrames.
metadata (pandas.Series): Metadata for samples. Index is sample names, values are class labels.
pathway_source (pandas.DataFrame): Pathway source data.
pathway_dict (dict): Dictionary of pathways. Keys are pathway names, values are lists of molecules.
sspa_scoring (object): Scoring method for SSPA.
min_coverage (int): Minimum number of omics required to cover a pathway.
sspa_method (object): SSPA scoring method.
sspa_scores_mv (dict): Dictionary of SSPA scores for each omics data. Keys are omics names, values are pandas DataFrames.
sspa_scores_sv (pandas.DataFrame): SSPA scores for all omics data concatenated.
coverage (dict): Dictionary of pathway coverage. Keys are pathway names, values are number of omics covering the pathway.
mv (object): Fitted MultiView model.
sv (object): Fitted SingleView model.
labels (pandas.Series): Class labels for samples. Index is sample names, values are class labels.
'''
def __init__(self, omics_data:dict, metadata, pathway_source, sspa_scoring=sspa.sspa_SVD, min_coverage=3):
self.omics_data = omics_data
self.omics_data_scaled = {k: pd.DataFrame(StandardScaler().fit_transform(v), columns=v.columns, index=v.index) for k, v in self.omics_data.items()}
self.metadata = metadata
self.pathway_source = pathway_source
self.pathway_dict = sspa.utils.pathwaydf_to_dict(pathway_source)
self.sspa_scoring = sspa_scoring
self.min_coverage = min_coverage
# sspa_methods = {'svd': sspa.sspa_SVD, 'ssGSEA': sspa.sspa_ssGSEA, 'kpca': sspa.sspa_KPCA, 'ssClustPA': sspa.sspa_ssClustPA, 'zscore': sspa.sspa_zscore}
self.sspa_method = self.sspa_scoring
self.sspa_scores_mv = None
self.sspa_scores_sv = None
self.coverage = self.get_multi_omics_coverage()
self.mv = None
self.sv = None
self.sv_us = None
self.labels = pd.factorize(self.metadata)[0]
def get_multi_omics_coverage(self):
all_molecules = sum([i.columns.tolist() for i in self.omics_data.values()], [])
coverage = {k: len(set(all_molecules).intersection(set(v))) for k, v in self.pathway_dict.items()}
return coverage
def MultiView(self, ncomp=2):
"""Fits a PathIntegrate MultiView model using MBPLS.
Args:
ncomp (int, optional): Number of components. Defaults to 2.
Returns:
object: Fitted PathIntegrate MultiView model.
"""
print('Generating pathway scores...')
sspa_scores_ = [self.sspa_method(self.pathway_source, self.min_coverage) for i in self.omics_data_scaled.values()]
sspa_scores = [sspa_scores_[n].fit_transform(i) for n, i in enumerate(self.omics_data_scaled.values())]
# sspa_scores = [self.sspa_method(self.pathway_source, self.min_coverage).fit_transform(i) for i in self.omics_data_scaled.values()]
# sspa_scores = [self.sspa_method(i, self.pathway_source, self.min_coverage, return_molecular_importance=True) for i in self.omics_data.values()]
self.sspa_scores_mv = dict(zip(self.omics_data.keys(), sspa_scores))
print('Fitting MultiView model')
mv = MBPLS(n_components=ncomp)
mv.fit([i.copy(deep=True) for i in self.sspa_scores_mv.values()], self.labels)
# compute VIP and scale VIP across omics
vip_scores = VIP_multiBlock(mv.W_, mv.Ts_, mv.P_, mv.V_)
vip_df = pd.DataFrame(vip_scores, index=sum([i.columns.tolist() for i in self.sspa_scores_mv.values()], []))
vip_df['Name'] = vip_df.index.map(dict(zip(self.pathway_source.index, self.pathway_source['Pathway_name'])))
vip_df['Source'] = sum([[k] * v.shape[1] for k, v in self.sspa_scores_mv.items()], [])
vip_df['VIP_scaled'] = vip_df.groupby('Source')[0].transform(lambda x: StandardScaler().fit_transform(x.values[:,np.newaxis]).ravel())
vip_df['VIP'] = vip_scores
mv.name = 'MultiView'
# only some sspa methods can return the molecular importance
if hasattr(sspa_scores_[0], 'molecular_importance'):
mv.molecular_importance = dict(zip(self.omics_data.keys(), [i.molecular_importance for i in sspa_scores_]))
mv.beta = mv.beta_.flatten()
mv.vip = vip_df
mv.omics_names = list(self.omics_data.keys())
mv.sspa_scores = self.sspa_scores_mv
mv.coverage = self.coverage
self.mv = mv
return self.mv
def SingleView(self, model=sklearn.linear_model.LogisticRegression, model_params=None):
"""Fits a PathIntegrate SingleView model using an SKLearn-compatible predictive model.
Args:
model (object, optional): SKlearn prediction model class. Defaults to sklearn.linear_model.LogisticRegression.
model_params (_type_, optional): Model-specific hyperparameters. Defaults to None.
Returns:
object: Fitted PathIntegrate SingleView model.
"""
concat_data = pd.concat(self.omics_data_scaled.values(), axis=1)
print('Generating pathway scores...')
sspa_scores = self.sspa_method(self.pathway_source, self.min_coverage)
self.sspa_scores_sv = sspa_scores.fit_transform(concat_data)
if model_params:
sv = model(**model_params) # ** this is inputed into the scikit learn model
else:
sv = model()
print('Fitting SingleView model')
# fitting the model
sv.fit(X=self.sspa_scores_sv, y=self.labels)
sv.sspa_scores = self.sspa_scores_sv
sv.name = 'SingleView'
sv.coverage = self.coverage
# only some sspa methods can return the molecular importance
if hasattr(sspa_scores, 'molecular_importance'):
sv.molecular_importance = sspa_scores.molecular_importance
self.sv = sv
return self.sv
# no cross validation in unsupervised (but can bootstrap)
# cross-validation approaches
def SingleViewClust(self, model=sklearn.cluster.KMeans,n_clusters_range=(2, 10), model_params=None, use_pca=True, pca_params=None, consensus_clustering=False, n_runs=10, auto_n_clusters=False, subsample_fraction=0.8, return_plot=False, return_ground_truth_plot=False, return_confusion_matrix=False, return_metrics_table=False):
"""
Fits a PathIntegrate SingleView Unsupervised model using an SKLearn-compatible KMeans model.
Args:
model (object, optional): SKLearn clustering model class. Defaults to sklearn.cluster.KMeans.
model_params (dict, optional): Model-specific hyperparameters. Defaults to None.
use_pca (bool, optional): Whether to perform PCA before clustering. Defaults to False.
pca_params (dict, optional): PCA-specific hyperparameters. Defaults to None.
consensus_clustering (bool, optional): Whether to perform consensus clustering. Defaults to False.
n_runs (int, optional): Number of runs for consensus clustering. Defaults to 10.
auto_n_clusters (bool, optional): Automatically determine the optimal number of clusters. Defaults to False.
n_clusters_range (tuple, optional): Range of cluster numbers to evaluate for optimal clusters. Defaults to (2, 10).
subsample_fraction (float, optional): Fraction of samples to use for each consensus clustering run. Defaults to 0.8.
return_plot (bool, optional): Whether to return a plot of the clustering result. Defaults to False.
return_ground_truth_plot (bool, optional): Whether to return a plot comparing the clustering result with ground truth. Defaults to False.
return_confusion_matrix (bool, optional): Whether to return a plot comparing different clustering algorithms. Defaults to False.
return_metrics_table (bool, optional): Whether to return a table of clustering evaluation metrics. Defaults to False.
Returns:
object: Fitted PathIntegrate SingleView Clustering model with various plots saved inside.
"""
# function to normalise the metrics later on
def normalize_score(score, score_min, score_max):
return (score - score_min) / (score_max - score_min)
# forming the concatenated data
concat_data = pd.concat(self.omics_data_scaled.values(), axis=1)
print('Generating pathway scores...')
sspa_scores = self.sspa_method(self.pathway_source, self.min_coverage)
self.sspa_scores_sv = sspa_scores.fit_transform(concat_data)
combined_data_scaled = StandardScaler().fit_transform(self.sspa_scores_sv)
combined_data_final = pd.DataFrame(combined_data_scaled, index=self.sspa_scores_sv.index, columns=self.sspa_scores_sv.columns)
self.sspa_scores_sv = combined_data_final
# if using PCA
if use_pca:
print('Performing PCA...')
if pca_params is None:
pca_params = {'n_components': min(concat_data.shape[1], 50)}
pca = sklearn.decomposition.PCA(**pca_params)
pca_components = pca.fit_transform(self.sspa_scores_sv)
component_names = [f'PC{i+1}' for i in range(pca_components.shape[1])]
self.sspa_scores_sv = pd.DataFrame(data=pca_components, columns=component_names, index=self.sspa_scores_sv.index)
else:
print('Not Using PCA...')
# if no model parameters
if model_params is None:
model_params = {}
# if automatically determining clusters
if auto_n_clusters:
print('Determining optimal number of clusters...')
best_score = -1
best_n_clusters = None
silhouette_scores = []
for n_clusters in range(*n_clusters_range):
sv_clust = model(n_clusters=n_clusters, **model_params)
labels = sv_clust.fit_predict(self.sspa_scores_sv)
silhouette_avg = sklearn.metrics.silhouette_score(self.sspa_scores_sv, labels)
silhouette_scores.append(silhouette_avg)
if silhouette_avg > best_score:
best_score = silhouette_avg
best_n_clusters = n_clusters
model_params['n_clusters'] = best_n_clusters
print(f'Optimal number of clusters determined: {best_n_clusters}')
# if using consensus clustering
# please provide subsample size
if consensus_clustering and n_runs > 0:
n_samples = self.sspa_scores_sv.shape[0]
consensus_matrix = np.zeros((n_samples, n_samples))
for run in range(n_runs):
print(f'Run {run + 1}/{n_runs}')
subsample_idx = np.random.choice(n_samples, int(subsample_fraction * n_samples), replace=False)
subsample_data = self.sspa_scores_sv.iloc[subsample_idx]
sv_clust = model(**(model_params or {}))
labels = sv_clust.fit_predict(subsample_data)
for i in range(len(subsample_idx)):
for j in range(i + 1, len(subsample_idx)):
if labels[i] == labels[j]:
consensus_matrix[subsample_idx[i], subsample_idx[j]] += 1
consensus_matrix[subsample_idx[j], subsample_idx[i]] += 1
consensus_matrix /= n_runs
consensus_labels = model(n_clusters=model_params['n_clusters']).fit_predict(consensus_matrix)
else:
sv_clust = model(**(model_params or {}))
consensus_labels = sv_clust.fit_predict(self.sspa_scores_sv)
# saving the variables in the object
self.sv_clust = sv_clust
self.sv_clust.sspa_scores = self.sspa_scores_sv
self.sv_clust.labels_ = consensus_labels
self.sv_clust.name = 'SingleViewClust'
print('Calculating clustering metrics...')
# various metrics caluclated
silhouette_avg = sklearn.metrics.silhouette_score(self.sspa_scores_sv, consensus_labels)
calinski_harabasz = sklearn.metrics.calinski_harabasz_score(self.sspa_scores_sv, consensus_labels)
davies_bouldin = sklearn.metrics.davies_bouldin_score(self.sspa_scores_sv, consensus_labels)
# normalising their scores using the previous function
silhouette_norm = normalize_score(silhouette_avg, -1, 1)
calinski_harabasz_norm = normalize_score(calinski_harabasz, 0, np.max([calinski_harabasz]))
davies_bouldin_norm = normalize_score(davies_bouldin, 0, np.max([davies_bouldin]))
davies_bouldin_norm = 1 - davies_bouldin_norm
combined_score = (silhouette_norm + calinski_harabasz_norm + davies_bouldin_norm) / 3
# saving the metrics
self.sv_clust.metrics = {
'Silhouette_Score': silhouette_avg,
}
# creating a clustering plot
if return_plot:
consensus_labels_series = pd.Series(consensus_labels, index=self.sspa_scores_sv.index, name='Consensus_Cluster')
sspa_scores_labels = self.sspa_scores_sv
sspa_scores_labels['Consensus_Cluster'] = consensus_labels_series
fig_clust = plt.figure(figsize=(10, 8))
sns.scatterplot(x=sspa_scores_labels.iloc[:, 0], y=sspa_scores_labels.iloc[:, 1], hue=sspa_scores_labels['Consensus_Cluster'], palette='coolwarm', s=100, edgecolor='black')
plt.title('Clustering Results', fontsize=20)
plt.xlabel('Component 1', fontsize=16)
plt.ylabel('Component 2', fontsize=16)
plt.xticks(fontsize=14)
plt.yticks(fontsize=14)
plt.legend(title='Cluster', title_fontsize=16, fontsize=14)
plt.grid(True)
plt.show()
sv_clust.clusters_plot = fig_clust
# creating a ground truth plot for comparison
if return_ground_truth_plot:
metadata_pca = self.metadata.to_frame(name='Who_Group')
sspa_scores_meta = pd.concat([self.sspa_scores_sv, metadata_pca], axis=1)
fig_ground = plt.figure(figsize=(10, 8))
sns.scatterplot(x=sspa_scores_meta.iloc[:, 0], y=sspa_scores_meta.iloc[:, 1], hue=sspa_scores_meta['Who_Group'], palette='coolwarm', s=100, edgecolor='black')
plt.title('Ground Truth Labels', fontsize=20)
plt.xlabel('Component 1', fontsize=16)
plt.ylabel('Component 2', fontsize=16)
plt.xticks(fontsize=14)
plt.yticks(fontsize=14)
plt.legend(title='Ground Truth', title_fontsize=16, fontsize=14)
plt.grid(True)
plt.show()
sv_clust.ground_truth_plot = fig_ground
# creating a confusion matrix to compare labels
if return_confusion_matrix:
metadata_pca = self.metadata.to_frame(name='Who_Group')
sspa_scores_meta = pd.concat([self.sspa_scores_sv, metadata_pca], axis=1)
consensus_labels_series = pd.Series(consensus_labels, index=sspa_scores_meta.index, name='Consensus_Cluster')
sspa_scores_meta['Consensus_Cluster'] = consensus_labels_series
confusion_df = pd.crosstab(sspa_scores_meta['Who_Group'], sspa_scores_meta['Consensus_Cluster'])
normalized_confusion_df = confusion_df.div(confusion_df.sum(axis=1), axis=0)
fig = go.Figure(data=go.Heatmap(
z=normalized_confusion_df.values,
x=normalized_confusion_df.columns,
y=normalized_confusion_df.index,
colorscale='Blues',
text=confusion_df.values,
texttemplate="%{text}",
hovertemplate="True Label: %{y}<br>Predicted: %{x}<br>Count: %{text}<extra></extra>",
colorbar=dict(title="Normalized Value")
))
fig.update_layout(
title=dict(
text="Confusion Matrix: Cluster vs Ground Truth Label",
font=dict(size=24)
),
xaxis_title=dict(
text="Predicted Cluster",
font=dict(size=18)
),
yaxis_title=dict(
text="Ground Truth Label",
font=dict(size=18)
),
xaxis=dict(
tickmode='array',
tickvals=list(range(len(confusion_df.columns))),
ticktext=confusion_df.columns,
tickfont=dict(size=14)
),
yaxis=dict(
tickmode='array',
tickvals=list(range(len(confusion_df.index))),
ticktext=confusion_df.index,
tickfont=dict(size=14)
),
height=600,
width=800
)
fig.show()
# calculating the ari score and adding this to the metrics (if confusion matrix is created)
ari_score = sklearn.metrics.adjusted_rand_score(sspa_scores_meta['Who_Group'], sspa_scores_meta['Consensus_Cluster'])
self.sv_clust.metrics['Adjusted_Rand_Index'] = ari_score
sv_clust.confusion_matrix = fig
# if the metrics table is requested
if return_metrics_table:
metrics_df = pd.DataFrame(self.sv_clust.metrics, index=[0])
metrics_df = metrics_df.T.reset_index()
metrics_df.columns = ['Metric', 'Value']
custom_palette = ['red' if i % 2 == 0 else 'blue' for i in range(len(metrics_df))]
fig_metrics = plt.figure(figsize=(10, 8))
ax = sns.barplot(y='Metric', x='Value', data=metrics_df, palette=custom_palette)
for index, value in enumerate(metrics_df['Value']):
plt.text(value / 2, index, f'{value:.2f}', color='white', ha='center', va='center', fontsize=18, weight='bold')
plt.ylabel('Metric', fontsize=18)
plt.xlabel('Value', fontsize=18)
plt.xticks(fontsize=18)
plt.yticks(fontsize=18)
plt.tight_layout()
plt.show()
sv_clust.metrics_plot = fig_metrics
print('Finished')
# creating a new sspa_scores obecjt with clusters
self.sv_clust.sspa_scores_clusters = self.sspa_scores_sv
return self.sv_clust
def SingleViewDimRed(self, model=sklearn.decomposition.PCA, model_params=None, return_pca_plot=False,return_tsne_plot = False, return_biplot=False, return_loadings_plot=False, return_tsne_density_plot=False ,metadata_continuous=False):
"""
Applies a dimensionality reduction technique to the input data.
Args:
model (object, optional): The dimensionality reduction model to use. Defaults to sklearn.decomposition.PCA.
model_params (dict, optional): Model-specific hyperparameters. Defaults to None.
return_pca_plot (bool, optional): Whether to return a PCA scatter plot of the first two principal components.
return_tsne_plot (bool, optional): Whether to return a t-SNE scatter plot of the first two components.
return_biplot (bool, optional): Whether to return a biplot (PCA plot with loadings).
return_loadings_plot (bool, optional): Whether to return a plot of the top loadings for each component.
return_tsne_density_plot (bool, optional): Whether to return a t-SNE scatter plot with a density overlay.
metadata_continuous (bool, optional): Whether metadata is continuous or categorical.
Returns:
object: Fitted dimensionality reduction model with reduced data and optional plots.
"""
# creating concatenated and data
concat_data = pd.concat(self.omics_data_scaled.values(), axis=1)
print('Generating pathway scores...')
sspa_scores = self.sspa_method(self.pathway_source, self.min_coverage)
self.sspa_scores_sv = sspa_scores.fit_transform(concat_data)
if model_params is None:
model_params = {}
sv_dim = model(**model_params) if model_params else model()
print('Fitting SingleView Dimensionality Reduction model')
# setging the model parameters depending on the model used (PCA or t-SNE)
if sklearn.decomposition.PCA:
if return_biplot or return_loadings_plot:
if model_params.get('n_components', 2) != 2:
print("Warning: n_components has been set to 2 for the biplot.")
model_params['n_components'] = 2
else:
model_params['n_components'] = 2
sv_dim = model(**model_params)
# scaling data and fitting the models
if model == sklearn.decomposition.PCA:
reduced_data_scaled = StandardScaler().fit_transform(self.sspa_scores_sv)
reduced_data_sspa = pd.DataFrame(reduced_data_scaled, columns=self.sspa_scores_sv.columns)
reduced_data = sv_dim.fit_transform(reduced_data_sspa)
explained_variance = sv_dim.explained_variance_ratio_ if hasattr(sv_dim, 'explained_variance_ratio_') else None
else:
reduced_data = sv_dim.fit_transform(self.sspa_scores_sv)
explained_variance = None
# saving to the dim object
sv_dim.reduced_data = reduced_data
sv_dim.explained_variance = explained_variance
sv_dim.sspa_scores_pca = self.sspa_scores_sv
sv_dim.name = 'SingleViewDimRed'
# incorporating metadata and setting it on a continuous scale if necessary
pca_df = pd.DataFrame(data=reduced_data[:, :2], columns=['PC1', 'PC2'])
metadata_pca = self.metadata.to_frame(name='Metadata').reset_index(drop=True)
if metadata_continuous:
metadata_pca['Meta_Group_Midpoint'] = metadata_pca['Metadata'].apply(self.convert_range_to_midpoint)
pca_df_named = pd.concat([pca_df, metadata_pca], axis=1)
pca_df_named = pca_df_named.sort_values('Meta_Group_Midpoint').reset_index(drop=True)
pca_df_named = pca_df_named.drop(columns=['Meta_Group_Midpoint'])
else:
pca_df_named = pd.concat([pca_df, metadata_pca], axis=1)
if return_tsne_plot or return_tsne_density_plot:
tsne_df = pd.DataFrame(data=reduced_data[:, :2], columns=['Component 1', 'Component 2'])
metadata_pca['Meta_Group_Midpoint'] = metadata_pca['Metadata'].apply(self.convert_range_to_midpoint)
tsne_df_named = pd.concat([tsne_df, metadata_pca], axis=1)
tsne_df_named = tsne_df_named.sort_values('Meta_Group_Midpoint').reset_index(drop=True)
tsne_df_named = tsne_df_named.drop(columns=['Meta_Group_Midpoint'])
# if returning tsne plot
if return_tsne_plot:
if model != sklearn.manifold.TSNE:
raise ValueError("Error: Please use sklearn.manifold.TSNE for t-SNE plots.")
sns.set_style("white")
fig_tsne = plt.figure()
sns.set_style("white")
sns.scatterplot(data=tsne_df_named, x='Component 1', y='Component 2', hue='Metadata', palette='coolwarm', s=100, edgecolor='black')
plt.title('t-SNE of Integrated Data (First 2 Components)')
plt.xlabel('Component 1')
plt.ylabel('Component 2')
plt.legend(title='Metadata Group')
plt.grid(True)
plt.show()
sv_dim.tsne_plot = fig_tsne
# if returning tsne density plot
if return_tsne_density_plot:
if model != sklearn.manifold.TSNE:
raise ValueError("Error: Please use sklearn.manifold.TSNE for t-SNE plots.")
sns.set_style("white")
fig_tsne_density = plt.figure(figsize=(10, 8))
sns.scatterplot(data=tsne_df_named, x='Component 1', y='Component 2', hue='Metadata', s=50, edgecolor='black', alpha=0.6, palette="coolwarm")
sns.kdeplot(data=tsne_df_named, x='Component 1', y='Component 2', hue='Metadata', fill=True, alpha=0.3, palette="coolwarm")
plt.title('t-SNE of Integrated Data with Density Overlay')
plt.xlabel('Component 1')
plt.ylabel('Component 2')
plt.legend(title='Metadata Group')
plt.grid(True)
plt.show()
sv_dim.tsne_density_plot = fig_tsne_density
# if a biplot or loadings plot is requested - need to convert pathways to names
if return_loadings_plot or return_biplot:
if model != sklearn.decomposition.PCA:
raise ValueError("Error: Please use sklearn.decomposition.PCA to examine loadings.")
loadings_df = pd.DataFrame(sv_dim.components_.T, columns=['Component 1', 'Component 2'], index=reduced_data_sspa.columns)
# function to download a kegg conversion file - reactome conversion file is already within code
url = "https://rest.kegg.jp/get/br:br08901/json"
response = requests.get(url)
if response.status_code == 200:
hierarchy_json = response.json()
with open('br08901.json', 'w') as f:
json.dump(hierarchy_json, f, indent=4)
else:
print("Failed to retrieve data. Status code:", response.status_code)
def create_id_name_mapping(node):
id_name_mapping = {}
if 'children' in node:
for child in node['children']:
id_name_mapping.update(create_id_name_mapping(child))
else:
pathway_id, pathway_name = node['name'].split(' ', 1)
id_name_mapping[pathway_id] = pathway_name.strip()
return id_name_mapping
pathway_mapping = create_id_name_mapping(hierarchy_json)
# extracting the top pathway loadings
top_loadings_pc1 = loadings_df['Component 1'].sort_values(key=abs, ascending=False).head(10)
top_loadings_pc2 = loadings_df['Component 2'].sort_values(key=abs, ascending=False).head(10)
top_loadings = pd.concat([top_loadings_pc1, top_loadings_pc2])
sv_dim.loadings_df = loadings_df
# creating a PCA plot
if return_pca_plot:
sns.set_style("white")
fig_pca = plt.figure()
sns.scatterplot(data=pca_df_named, x='PC1', y='PC2', hue='Metadata', palette="coolwarm", s=100, edgecolor='black')
plt.title('PCA of Integrated Data (First 2 Principal Components)')
plt.xlabel('Principal Component 1')
plt.ylabel('Principal Component 2')
plt.legend(title='Metadata Group')
plt.grid(True)
plt.show()
sv_dim.pca_plot = fig_pca
# creatign a biplot
if return_biplot:
scaling_factor = 200
sns.set_style("white")
fig_biplot = plt.figure(figsize=(10, 8))
sns.scatterplot(data=pca_df_named, x='PC1', y='PC2', hue='Metadata', s=100, edgecolor='black', alpha=0.2, legend=False)
for variable in top_loadings.index:
color = 'red' if variable in top_loadings_pc1.index else 'blue'
plt.arrow(0, 0, loadings_df.loc[variable, 'Component 1'] * scaling_factor,
loadings_df.loc[variable, 'Component 2'] * scaling_factor,
color=color, alpha=0.8, head_width=0.5, linewidth=2)
plt.text(loadings_df.loc[variable, 'Component 1'] * scaling_factor * 1.15,
loadings_df.loc[variable, 'Component 2'] * scaling_factor * 1.15,
variable, color='black', ha='center', va='center', fontsize=10)
plt.xticks(fontsize=14)
plt.yticks(fontsize=14)
plt.title('Biplot of PCA with Top 10 Loadings Highlighted')
plt.xlabel('Principal Component 1')
plt.ylabel('Principal Component 2')
plt.grid(True)
plt.show()
sv_dim.biplot = fig_biplot
# creating a loadings plot
if return_loadings_plot:
fig_loadings_plot = plt.figure(figsize=(12, 10))
index_mapping = self.pathway_source['Pathway_name'].to_dict()
def rename_index(index):
if index.startswith('R-HSA'):
return index_mapping.get(index, index)
else:
return pathway_mapping.get(index, index)
loadings_df.index = [rename_index(idx) for idx in loadings_df.index]
top_loadings_pc1 = loadings_df['Component 1'].sort_values(key=abs, ascending=False).head(10)
top_loadings_pc2 = loadings_df['Component 2'].sort_values(key=abs, ascending=False).head(10)
top_loadings = pd.concat([top_loadings_pc1, top_loadings_pc2])
colors = ['red' if variable in top_loadings_pc1.index else 'blue' for variable in top_loadings.index]
top_loadings.plot(kind='barh', color=colors, width=0.3)
plt.title('Top 5 Loadings for PC1 and PC2', fontsize=24)
plt.xlabel('Loading Value', fontsize=20)
plt.ylabel('', fontsize=30)
plt.axvline(0, color='black', linewidth=0.1)
plt.xticks(rotation=45, fontsize=18)
plt.yticks(fontsize=30)
plt.show()
sv_dim.loadings_plot = fig_loadings_plot
sv_dim.metadata_pca = metadata_pca
self.sv_dim = sv_dim
return self.sv_dim
# function to convert ranges to midpoints
def convert_range_to_midpoint(self, value):
"""
Converts a range like '10-20' into its midpoint value '15'.
"""
if isinstance(value, str) and '-' in value:
try:
start, end = map(float, value.split('-'))
return (start + end) / 2
except ValueError:
return value
return value
def SingleViewGridSearchCV(self, param_grid, model=sklearn.linear_model.LogisticRegression, grid_search_params=None):
'''Grid search cross-validation for SingleView model.
Args:
param_grid (dict): Grid search parameters.
model (object, optional): SKlearn prediction model class. Defaults to sklearn.linear_model.LogisticRegression.
grid_search_params (dict, optional): Grid search parameters. Defaults to None.
Returns:
object: GridSearchCV object.
'''
# concatenate omics - unscaled to avoid data leakage
concat_data = pd.concat(self.omics_data.values(), axis=1)
# Set up sklearn pipeline
pipe_sv = sklearn.pipeline.Pipeline([
('Scaler', StandardScaler().set_output(transform="pandas")),
('sspa', self.sspa_method(self.pathway_source, self.min_coverage)),
('model', model())
])
# Set up cross-validation
grid_search = GridSearchCV(pipe_sv, param_grid=param_grid, **grid_search_params)
grid_search.fit(X=concat_data, y=self.labels)
return grid_search
# only 1 model so one parameter way to grid search pca components
# advantage of multi view is interpretation of contribution
def MultiViewCV(self):
'''Cross-validation for MultiView model.
Returns:
object: Cross-validation results.
'''
# Set up sklearn pipeline
pipe_mv = sklearn.pipeline.Pipeline([
('sspa', self.sspa_method(self.pathway_source, self.min_coverage)),
('mbpls', MBPLS(n_components=2))
])
# Set up cross-validation
cv_res = cross_val_score(pipe_mv, X=[i.copy(deep=True) for i in self.omics_data.values()], y=self.labels)
return cv_res
def MultiViewGridSearchCV(self):
pass
def VIP_multiBlock(x_weights, x_superscores, x_loadings, y_loadings): “"”Calculate VIP scores for multi-block PLS.
Args:
x_weights (list): List of x weights.
x_superscores (list): List of x superscores.
x_loadings (list): List of x loadings.
y_loadings (list): List of y loadings.
Returns:
numpy.ndarray: VIP scores for each feature across all blocks. Features are in original order.
"""
# stack the weights from all blocks
weights = np.vstack(x_weights)
# calculate product of sum of squares of superscores and y loadings
sumsquares = np.sum(x_superscores**2, axis=0) * np.sum(y_loadings**2, axis=0)
# p = number of variables - stack the loadings from all blocks
p = np.vstack(x_loadings).shape[0]
# VIP is a weighted sum of squares of PLS weights
vip_scores = np.sqrt(p * np.sum(sumsquares*(weights**2), axis=1) / np.sum(sumsquares))
return vip_scores
Recommended citation: Popham, J., Wieder, C., & Ebbels, T. (2024). cwieder/PathIntegrate: PathIntegrate Unsupervised (v1.0.0). Zenodo. --> 'Wieder C, Cooke J, Frainay C, et al. (2024). PathIntegrate: Multivariate Modelling Approaches for Pathway-Based Multi-Omics Data Integration. PLOS Computational Biology. 20(3): e1011814. doi:10.1371/journal.pcbi.1011814.
Download Paper